[Tempo di lettura: 10 min]
 La comprensione di semplici concetti di farmacocinetica è molto utile nella pratica, per stabilire l'intervallo di somministrazione di un farmaco, sapere quando aspettarsi l'inizio dell'effetto o la comparsa di effetti avversi e prevederne la scomparsa dopo la sospensione. [Lettura 8 min]
La comprensione di semplici concetti di farmacocinetica è molto utile nella pratica, per stabilire l'intervallo di somministrazione di un farmaco, sapere quando aspettarsi l'inizio dell'effetto o la comparsa di effetti avversi e prevederne la scomparsa dopo la sospensione. [Lettura 8 min]
I concetti della farmacocinetica possono sembrare astrusi, ma, che ci si pensi o meno, influenzano la nostra pratica prescrittiva quotidiana.
La farmacocinetica studia le modalità con cui i farmaci entrano nell'organismo, vengono distribuiti, metabolizzati ed eliminati.
Conoscere approssimativamente il tempo in cui un farmaco raggiunge la concentrazione massima nel sangue (Tmax) aiuta a prevedere la comparsa degli effetti di farmaci che agiscono rapidamente.
Conoscere il tempo medio di dimezzamento della concentrazione del farmaco (T½) può definire una durata minima per i tentativi terapeutici.
La conoscenza dell'emivita ci informa anche sul momento in cui ci si devono aspettare sintomi di astinenza dopo la sospensione della terapia.
Tmax e comparsa dell’effetto farmacologico - Il Tmax è il tempo necessario per raggiungere il picco di concentrazione nel sangue dopo la somministrazione di un farmaco.
Nella Figura, la freccia blu mostra il Tmax dopo una dose di un ipotetico farmaco.
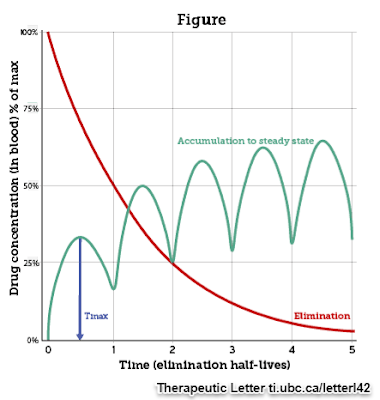
Per i farmaci somministrati per via orale, il Tmax può essere influenzato dall'assunzione a digiuno o dopo un pasto e dal ritardo nello svuotamento dello stomaco.
I farmaci somministrati tramite bolo endovenoso raggiungono il Tmax quasi istantaneamente, e alcuni farmaci inalati si comportano in modo simile (p.es. ossigeno, nicotina, cannabis).
Emivita (T½) - È il tempo medio in cui la concentrazione di un farmaco si dimezza dopo aver raggiunto il picco nel sangue.
Il T½ può avere molta variabilità, ma la media può essere utile per prevedere la durata degli effetti della maggior parte dei farmaci.
Qualsiasi cosa influenzi distribuzione, metabolismo ed escrezione può aumentare l'emivita.

I concetti della farmacocinetica possono sembrare astrusi, ma, che ci si pensi o meno, influenzano la nostra pratica prescrittiva quotidiana.
La farmacocinetica studia le modalità con cui i farmaci entrano nell'organismo, vengono distribuiti, metabolizzati ed eliminati.
Conoscere approssimativamente il tempo in cui un farmaco raggiunge la concentrazione massima nel sangue (Tmax) aiuta a prevedere la comparsa degli effetti di farmaci che agiscono rapidamente.
Conoscere il tempo medio di dimezzamento della concentrazione del farmaco (T½) può definire una durata minima per i tentativi terapeutici.
La conoscenza dell'emivita ci informa anche sul momento in cui ci si devono aspettare sintomi di astinenza dopo la sospensione della terapia.
Tmax e comparsa dell’effetto farmacologico - Il Tmax è il tempo necessario per raggiungere il picco di concentrazione nel sangue dopo la somministrazione di un farmaco.
Nella Figura, la freccia blu mostra il Tmax dopo una dose di un ipotetico farmaco.

Per i farmaci somministrati per via orale, il Tmax può essere influenzato dall'assunzione a digiuno o dopo un pasto e dal ritardo nello svuotamento dello stomaco.
I farmaci somministrati tramite bolo endovenoso raggiungono il Tmax quasi istantaneamente, e alcuni farmaci inalati si comportano in modo simile (p.es. ossigeno, nicotina, cannabis).
Emivita (T½) - È il tempo medio in cui la concentrazione di un farmaco si dimezza dopo aver raggiunto il picco nel sangue.
Il T½ può avere molta variabilità, ma la media può essere utile per prevedere la durata degli effetti della maggior parte dei farmaci.
Qualsiasi cosa influenzi distribuzione, metabolismo ed escrezione può aumentare l'emivita.
Per quanto riguarda la distribuzione, per farmaci come il diazepam che si distribuiscono nel tessuto adiposo, una maggiore quantità di adipe è associata a un maggiore immagazzinamento e quindi a una maggiore disponibilità del farmaco che ritorna nel sangue quando i livelli ematici si riducono.
Il risultato netto è un'emivita più lunga. Gli anziani tendono ad avere un maggior rapporto massa grassa/massa magra e questo è uno dei motivi per cui l'emivita di alcuni farmaci, come il diazepam, è maggiore.
Relativamente al metabolismo, gli individui ereditano diverse predisposizioni metaboliche.
Come accennato nel post sul citocromo 450, il CYP2D6 è l’isoforma più soggetta a polimorfismo.
La maggior parte delle persone (67%-90%) sono metabolizzatori rapidi del CYP2D6; il resto, a seconda dell'etnia, sono metabolizzatori lenti, intermedi o ultrarapidi.
Quindi, l'emivita dei numerosi farmaci metabolizzati dal CYP2D6 (p.es. codeina, tramadolo, risperidone, venlafaxina) è molto influenzata dallo stato metabolico individuale.
L'emivita di eliminazione è piuttosto rilevante per le decisioni cliniche.
Predice la durata degli effetti per farmaci assunti per via orale e per quelli somministrati per via endovenosa dopo la redistribuzione.
Se la dose somministrata di un farmaco è maggiore della dose eliminata tra una dose e l'altra, sono necessarie circa 5 emivite perché la quantità di farmaco assunta ogni giorno sia uguale alla quantità eliminata (steady state).

I livelli ematici allo steady state sono maggiori o minori quando i farmaci vengono somministrati più o meno frequentemente.
Lo steady state è importante perché è il momento in cui il livello ematico è ragionevolmente stabile tra una dose e la successiva.
Per quei farmaci di cui è importante il monitoraggio terapeutico (p.es. valproato, levetiracetam, litio, clozapina), i livelli ematici vanno controllati solo dopo aver raggiunto lo steady state.
È importante notare che, mentre alcuni effetti collaterali possono emergere precocemente a causa dell'improvvisa esposizione dell'organismo a una nuova sostanza, gli effetti avversi tendono a essere chiaramente evidenti solo quando viene raggiunta una soglia critica, di solito correlata al livello ematico.
I livelli ematici non sono uniformi allo steady state: raggiungono un picco transitorio quando il farmaco viene assorbito dopo ogni somministrazione e poi diminuiscono gradualmente, fino a raggiungere un valore minimo poco prima della somministrazione successiva.
Allo steady state picchi e valli non sono molto distanti tra loro, in termini di concentrazione. Quanto più è breve l'intervallo tra una dose e l'altra, tanto minore è la differenza nei livelli ematici tra picco e valle.
Se, per esempio, la carbamazepina deve essere somministrata con un dosaggio di 600 mg/d, le fluttuazioni dei livelli ematici tra picchi e valli saranno minori assumendo 200 mg tre volte al giorno rispetto a una singola dose di 600 mg alla sera.
Se la dose somministrata di un farmaco è maggiore della dose eliminata tra una dose e l'altra, sono necessarie circa 5 emivite perché la quantità di farmaco assunta ogni giorno sia uguale alla quantità eliminata (steady state).

I livelli ematici allo steady state sono maggiori o minori quando i farmaci vengono somministrati più o meno frequentemente.
Lo steady state è importante perché è il momento in cui il livello ematico è ragionevolmente stabile tra una dose e la successiva.
Per quei farmaci di cui è importante il monitoraggio terapeutico (p.es. valproato, levetiracetam, litio, clozapina), i livelli ematici vanno controllati solo dopo aver raggiunto lo steady state.
È importante notare che, mentre alcuni effetti collaterali possono emergere precocemente a causa dell'improvvisa esposizione dell'organismo a una nuova sostanza, gli effetti avversi tendono a essere chiaramente evidenti solo quando viene raggiunta una soglia critica, di solito correlata al livello ematico.
I livelli ematici non sono uniformi allo steady state: raggiungono un picco transitorio quando il farmaco viene assorbito dopo ogni somministrazione e poi diminuiscono gradualmente, fino a raggiungere un valore minimo poco prima della somministrazione successiva.
Allo steady state picchi e valli non sono molto distanti tra loro, in termini di concentrazione. Quanto più è breve l'intervallo tra una dose e l'altra, tanto minore è la differenza nei livelli ematici tra picco e valle.
Se, per esempio, la carbamazepina deve essere somministrata con un dosaggio di 600 mg/d, le fluttuazioni dei livelli ematici tra picchi e valli saranno minori assumendo 200 mg tre volte al giorno rispetto a una singola dose di 600 mg alla sera.
Allo steady state i picchi possono essere associati a effetti avversi, mentre le riduzioni possono risultare in una perdita di efficacia.
Per questo motivo, il dosaggio ematico va valutato durante le fasi di riduzione: il prelievo di sangue va fatto poco prima della somministrazione successiva.
I livelli di picco sono meno accurati per il monitoraggio terapeutico a causa della variabilità individuale e sono riservati a farmaci con emivita breve in cui i livelli di picco sono associati all'efficacia o alla tossicità, per esempio la gentamicina.
I valori di Tmax e T½ indicati nelle schede tecniche sono valori medi ottenuti su piccoli gruppi.
Esistono tuttavia importanti differenze interindividuali che possono dipendere dal polimorfismo genetico e da altri fattori, come la riduzione della funzione renale.
Quando un paziente ci dice che l'effetto di un farmaco si esaurisce più velocemente o dura molto più a lungo del previsto, il suo T½ individuale può differire dalla media riportata in scheda tecnica.
Se, per esempio, un paziente ci dice che l'effetto analgesico di un farmaco si esaurisce molto prima dell'assunzione successiva, vanno considerati i parametri farmacocinetici: se il farmaco raggiunge rapidamente il Tmax ma l'emivita è breve, da 1 a 4 ore, si può considerare un'assunzione più frequente o una formulazione con assorbimento ritardato.
Eliminazione - La maggior parte dei farmaci viene eliminata attraverso il metabolismo epatico e/o l'escrezione renale.
Quando è proporzionale alla concentrazione ematica, si parla di cinetica di "primo ordine".
Una volta che un farmaco viene sospeso, la concentrazionesi riduce in modo esponenziale (linea rossa nella figura in alto a destra).
Il valore di T½ suggerisce quando i livelli ematici saranno trascurabili. Va tenuto presente che la variabilità interindividuale del T½ e una ridotta funzionalità renale o un'epatopatia grave possono prolungare di molto le tempistiche.
Fanno eccezione i farmaci che vengono eliminati a una velocità fissa, quando la loro concentrazione satura le vie metaboliche (p.es. alcol, fenitoina, paracetamolo). In questi casi si parla di cinetica di "ordine zero".
Per i farmaci con cinetica di ordine zero, viene eliminata una quantità costante di farmaco nell'unità di tempo. L'emivita non è costante, ma dipende dalla concentrazione.
In caso di sovradosaggio da farmaci con cinetica di ordine zero, la somministrazione va interrotta immediatamente: solo dopo la riduzione della concentrazione ematica gli effetti del farmaco diminuiscono.
In pratica - Conoscere il Tmax può essere utile per stabilire quando valutare gli effetti di un farmaco (positivi o negativi).
Sono necessarie 4-5 emivite per valutare gli effetti farmacologici allo steady state.
Quando un farmaco viene sospeso, è necessario un intervallo di tempo simile perché gli effetti scompaiano o emergano potenziali sintomi di astinenza.
L'emivita indicata in scheda tecnica è una media, ma ci possono essere significative differenze interindividuali.
I dati di farmacocinetica sono reperibili nella relativa sezione delle schede tecniche dei farmaci.
Una fonte di informazioni utile è il database Micromedex consultabile gratuitamente, per esempio, tramite la Biblioteca Virtuale per la Salute del Piemonte.
Il Micromedex Assistant è una funzione utile per la consultazione rapida.
Per esempio, dovendo somministrare del fluconazolo a un paziente che assume anche ranolazina (associazione controindicata) è utile sospendere (se possibile) la ranolazina durante la terapia con fluconazolo.
Inserendo su Micromedex PK FLUCONAZOLE si ottengono i dati di farmacocinetica, tra cui l’emivita di eliminazione (30 ore nell’adulto, 46 nell’anziano).
Dopo l’ultima somministrazione del fluconazolo, quindi, è prudente attendere almeno un giorno (anche due nell’anziano) prima di riprendere l’assunzione della ranolazina.
[142] Simple clinical pharmacology can improve prescribing
Therapeutics Initiative - 29 May 2023
The Practical Importance of Half-Life in Psychopharmacology
J Clin Psychiatry. 2022 Jul 25;83(4):22f14584
Practical pharmacokinetics: what do you really need to know?
Arch Dis Child Educ Pract Ed. 2015 Feb;100(1):37-43
L'emivita indicata in scheda tecnica è una media, ma ci possono essere significative differenze interindividuali.
I dati di farmacocinetica sono reperibili nella relativa sezione delle schede tecniche dei farmaci.
Una fonte di informazioni utile è il database Micromedex consultabile gratuitamente, per esempio, tramite la Biblioteca Virtuale per la Salute del Piemonte.

Il Micromedex Assistant è una funzione utile per la consultazione rapida.
Per esempio, dovendo somministrare del fluconazolo a un paziente che assume anche ranolazina (associazione controindicata) è utile sospendere (se possibile) la ranolazina durante la terapia con fluconazolo.
Inserendo su Micromedex PK FLUCONAZOLE si ottengono i dati di farmacocinetica, tra cui l’emivita di eliminazione (30 ore nell’adulto, 46 nell’anziano).
Dopo l’ultima somministrazione del fluconazolo, quindi, è prudente attendere almeno un giorno (anche due nell’anziano) prima di riprendere l’assunzione della ranolazina.
[142] Simple clinical pharmacology can improve prescribing
Therapeutics Initiative - 29 May 2023
The Practical Importance of Half-Life in Psychopharmacology
J Clin Psychiatry. 2022 Jul 25;83(4):22f14584
Practical pharmacokinetics: what do you really need to know?
Arch Dis Child Educ Pract Ed. 2015 Feb;100(1):37-43
Gilberto Lacchia - Pubblicato 05/06/2023 - Aggiornato 05/06/2023
Commenti
Posta un commento